В клеточная лимфома крупноклеточная, диффузная: шансы на выживание, прогноз
В настоящее время к проблемам неходжкинских лимфом (НХЛ) привлечено внимание исследователей самых различных специальностей: гематологов, онкологов, терапевтов, иммунологов, морфологов и др. Это связано с неуклонным ростом заболеваемости, особенно в развитых странах, где она за последние 20 лет увеличилась более чем на 50% и по темпу прироста превышает лимфому Ходжкина. НХЛ представляют собой гетерогенную группу злокачественных лимфопролиферативных опухолей, различающихся по биологическим свойствам, морфологическому строению, клиническим проявлениям, ответу на терапию и прогнозу. Биологическая и морфологическая гетерогенность НХЛ до сих пор создает значительные трудности как для первичной диагностики, так и для выработки адекватного плана лечения.
При НХЛ 5-летняя выживаемость широко варьирует в зависимости от морфоиммунологического варианта опухоли: при В-клеточных лимфомах маргинальной зоны, MALT-лимфомах и фолликулярных лимфомах она превышает 70%, что трактуется как очень хороший прогноз, тогда как при Т-лимфобластных, периферических Т-клеточных НХЛ этот показатель ниже 30% [1]. Условно НХЛ можно разделить на две группы — лимфомы высокой и низкой степени злокачественности.
Для НХЛ высокой степени злокачественности (диффузные крупноклеточные лимфомы, лимфома Беркитта, лимфобластные лимфомы и т.д.) характерно агрессивное течение, без противоопухолевой терапии быстро приводящее больного к смерти. В то же время адекватная противоопухолевая терапия позволяет в ряде случаев излечивать пациентов. НХЛ низкой степени злокачественности (индолентные лимфомы) в ряде случаев даже без противоопухолевой терапии могут протекать годами, не доставляя пациенту выраженного дискомфорта. Для таких пациентов даже существует тактика «наблюдай и жди», при которой противоопухолевое лечение начинают только в случае появления симптомов опухолевого роста, нарушающих качество жизни больных. Однако ни один из видов цитостатической терапии, известной в настоящее время, не способен излечить пациентов с развернутой стадией индолентных НХЛ. Проведение химиотерапии больным с лимфомами низкой степени злокачественности позволяет достаточно эффективно купировать симптомы и даже достигать полной регрессии видимых проявлений болезни, однако в дальнейшем у всех больных наблюдаются прогрессирование/рецидивы заболевания. При развитии рецидива шанс на достижение последующей ремиссии уменьшается, а длительность повторных ремиссий становится все меньше. Таким образом, разделение всех вариантов опухоли по степени злокачественности остается наиболее важным для клиницистов, так как позволяет разграничить подходы к терапии: при агрессивных НХЛ — максимально агрессивное лечение, целью которого является излечение больного, при индолентных — терапия хронического заболевания, нацеленная на продление жизни и сохранение ее качества.
Большое внимание уделяется прогностической ценности различных клинико-лабораторных характеристик заболевания. На основании тщательного изучения всех проявлений НХЛ было установлено, что наиболее значимыми факторами неблагоприятного прогноза являются возраст старше 60 лет, повышение уровня ЛДГ, общее состояние больного, соответствующее II—IV степени (ECOG), III—IV стадия болезни, наличие более одного экстранодального очага поражения. Все эти показатели вошли в так называемый международный прогностический индекс (МПИ), определение которого в настоящее время является стандартным при оценке прогноза для пациента. Недавно подтверждено негативное влияние на прогноз анемии, гипопротеинемии и гипоальбуминемии.
Для лечения НХЛ применяются все виды противоопухолевой терапии. Основными факторами, влияющими на выбор терапевтической тактики, являются морфоиммунологический вариант опухоли, преимущественная локализация опухолевого процесса и факторы прогноза (МПИ и т.д.). Учитывая биологические особенности различных вариантов НХЛ, при выборе лечебной тактики следует помнить, что при агрессивных лимфомах основной задачей является достижение полной ремиссии, ибо даже в тех случаях, когда удается достичь лишь частичной ремиссии, 5-летняя выживаемость пациентов в 3 раза меньше (50 и 15% соответственно). В отличие от агрессивных лимфом при индолентных лимфомах 5-летняя выживаемость может превышать 80% независимо от эффективности лечения. Наиболее распространенным вариантом индолентных зрелоклеточных НХЛ является фолликулярная лимфома — моноклональная опухоль из зрелых В-лимфоидных клеток, составляющая 30—40% всех лимфоидных опухолей взрослых. Прогноз в целом благоприятный: 5-летняя выживаемость равна 70%, 10-летняя — 15%.
Морфологически фолликулярная лимфома состоит или из преимущественно мелких В-лимфоидных клеток с извитым ядром (центроциты), или из их сочетания с крупными клетками (центробласты). Прогноз значительно ухудшается при увеличении в опухолевой ткани количества крупных клеток. Это легло в основу морфологического выделения трех типов фолликулярной НХЛ: I тип — опухоль состоит преимущественно из мелких клеток, II тип — смешанное представительство мелких и крупных клеток, III тип — преобладание в опухолевой ткани крупных клеток. Морфологическая разнородность фолликулярной лимфомы и связанная с этим различная чувствительность к терапии, а также возможность трансформации этого варианта в более агрессивные формы определяют принципиально отличающиеся друг от друга терапевтические подходы при каждом ее типе: от выжидательной тактики до высокоинтенсивного лечения.
В лечении фолликулярной лимфомы используются все виды терапии: лучевая, монохимиотерапия алкилирующими агентами, полихимиотерапия, иммунотерапия (интерферон-а, моноклональные антитела), однако оптимальные подходы не определены до сих пор. Возможность наступления самопроизвольных ремиссий (у 25% больных) позволила обосновать лечебную тактику «наблюдай и жди», заключающуюся в тщательном динамическом наблюдении за этими больными и начале противоопухолевой терапии только в случае возникновения симптомов, обусловленных прогрессированием опухоли. Алкилирующие агенты (хлорамбуцил и циклофосфан) при благоприятном течении фолликулярной лимфомы I типа могут с успехом использоваться в монорежиме довольно длительное время. Наличие неблагоприятных факторов при фолликулярной НХЛ I типа и фолликулярная лимфома II типа независимо от выраженности клинических признаков являются показанием к использованию полихимиотерапии в качестве I линии лечения (схемы LVPP, COP, CVP). Комбинация CVP (циклофосфан, винкристин, преднизолон) стала стандартом лечения во многих европейских центрах. Однако общая тенденция лечения индолентных лимфом в последнее время заключается в интенсификации I линии терапии. Это основано на возможности достижения у большего числа больных полных и более продолжительных ремиссий. Использование антрациклинов в I линии лечения при наличии неблагоприятных факторов прогноза и III типе опухоли обеспечивает 3-летнюю медиану безрецидивной выживаемости.
Значительным достижением в лечении НХЛ этой группы больных можно считать появившуюся возможность использования моноклональных антител к антигену CD-20 — ритуксимаба (мабтера), проявивших в клинических исследованиях противоопухолевую эффективность в сочетании с безопасностью при CD-20-позитивных лимфопролиферативных процессах. Основными показаниями к использованию ритуксимаба являются индолентные НХЛ. Первые позитивные результаты были получены в 1998—1999 гг. при лечении 166 больных с рецидивами и рефрактерными формами индолентных НХЛ. Эффект при использовании мабтеры в монорежиме был получен у 48% пациентов, при этом количество полных ремиссий было невелико — 4%, тем не менее достигнутые ремиссии оказались весьма стойкими — средняя продолжительность ответа составила 11,8 мес [2]. Заслуживают внимания данные Т.A. Davis (1999), который подтвердил целесообразность и безопасность повторного использования мабтеры при развитии рецидива заболевания после ранее проведенной успешной терапии моноклональными антителами — почти у половины (44%) больных вновь удается добиться успеха (полная ремиссия — 11%, частичная ремиссия — 33%, медиана полных ремиссий — 4,8 мес). Это подкрепляется сведениями о минимальной частоте развития антител к мабтере, что позволяет с успехом планировать повторное лечение [3].
Позднее была показана эффективность мабтеры в режиме монотерапии у первичных больных индо-лентными НХЛ. Общая эффективность достигла 73%, полные ремиссии были получены у 26% больных, время до прогрессирования составило почти 2 года. В настоящее время обосновано мнение о целесообразности использования мабтеры в сочетании с полихимиотерапией в I линии терапии III—IV стадий фолликулярной лимфомы. В 2003 г. были опубликованы результаты крупного рандомизированного исследования по сравнительному изучению схем CVP и CVP в сочетании с мабтерой (R-CVP) у 322 больных с III—IV стадиями фолликулярной лимфомы в I линии терапии. При сравнительной оценке оказалось, что 8 циклов R-CVP увеличили частоту общих ответов до 81% в сравнении с 57% в группе СVP (р
Диффузная В-крупноклеточная лимфома
Диффузная В крупноклеточная лимфома (ДВКЛ) — это группа опухолей лимфатической системы, в основе которой лежит трансформация В-лимфоцитов в злокачественные клетки.
- Причины возникновения и группы риска
- Симптомы диффузной крупноклеточной В лимфомы
- Классификация, формы и стадии диффузной лимфомы
- Диагностика диффузной крупноклеточной лимфомы
- Лечение диффузной В-крупноклеточной лимфомы
- Восстановление после лечения крупноклеточной неходжкинской лимфомы
- Рецидивы крупноклеточной лимфомы
- Осложнения диффузной В крупноклеточной лимфомы
- Прогноз и профилактика крупноклеточной лимфомы
Причины возникновения и группы риска
Причиной возникновения ДВКЛ является изменение В-лимфоцитов, т. е. нарушение структуры ДНК. Из-за этого клетки начинают бесконтрольно расти и размножаться, рассеиваясь по всему организму и поражая различные органы и системы. Почему возникают эти мутации, достоверно не установлено. Более того, есть много людей, у которых обнаруживаются характерные генетические поломки, но нет лимфомы. Данный вопрос продолжает изучаться.
В настоящее время принято говорить о факторах риска, которые повышают вероятность развития данного заболевания:
- Врожденные и приобретенные иммунодефициты: синдром Вискотта-Олдрича, Луи-Бара, СПИД, необходимость лечения, которое снижает иммунитет: цитостатики, иммуносупрессоры при трансплантации органов и др.
- Вирусные инфекции: ВИЧ, гепатит С, Т-лимфотропный вирус.
- Аутоиммунные заболевания: ревматоидный артрит, волчанка и др.
- Токсическое воздействие инсектицидов, гербицидов, бензола.
- Лечение посредством цитостатической и лучевой терапии.
Симптомы диффузной крупноклеточной В лимфомы
Крупноклеточная лимфома может проявляться множеством симптомов, но все их многообразие можно уложить в три синдрома:
- Увеличение лимфатических узлов — лимфаденопатия. Чаще всего, лимфома манифестирует безболезненным увеличением лимфатических узлов, которые можно обнаружить визуально или при пальпации. Но бывает так, что поражаются узлы, недоступные для осмотра и пальпации (например, в грудной или брюшной полости), тогда этого признака сразу может и не быть.
- Симптомы интоксикации: повышение температуры, не связанное с инфекцией, сильная потливость, потеря веса. Комбинацию этих признаков называют В-симптоматикой. Ее наличие имеет значение при определении стадии заболевания.
- Симптомы, которые развиваются, когда крупноклеточная лимфома переходит на другие органы и системы. Это могут быть боли в груди и кашель, боли в животе и нарушение стула. При поражении ЦНС развиваются головные боли, нарушение зрения и рвота. Из-за разрушения костного мозга снижается иммунитет, развивается анемия и склонность к опасным кровотечениям.
Классификация, формы и стадии диффузной лимфомы
Современная классификация крупноклеточной лимфомы базируется на клинических данных и степени распространенности опухолевого процесса.
Классификация Ann Arbor:
- 1 стадия. Поражение ограничено одной лимфатической зоной или одним экстралимфатическим органом или одним сегментом экстралимфатической ткани.
- 2 стадия. Поражено две и более зоны лимфатических узлов по одну сторону диафрагмы. При поражении внелимфатических органов или тканей обнаруживаются метастазы в регионарные лимфатические узлы.
- 3 стадия. Имеются единичные очаги поражения по обе стороны диафрагмы.
- 4 стадия. Имеются множественные очаги поражения экстралимфатических органов, либо ограниченное поражение экстралимфатических органов с отдаленными метастазами, либо поражение печени и костного мозга.

Классификация Ann Arbor дополняется модификацией Cotswold:
- А — нет В-симптомов.
- В — есть хотя бы один из В-симптомов.
- Е — есть локализованные экстранодальные очаги.
- S — лимфома поразила селезенку.
- X — имеется массивное опухолевое тканей и внутренних органов.
Диагностика диффузной крупноклеточной лимфомы
Диагноз неходжкинская лимфома выставляется на основании гистологического и иммуногистохимического исследования опухолевой ткани. Для того чтобы получить материал, проводят инцизионную или эксцизионную биопсию лимфатических узлов. Помимо этого, выполняют молекулярно-генетические и иммунофенотипические исследования, которые позволят определить вид опухоли, наличие тех или иных генетических изменений и подобрать оптимальный метод лечения.
Для определения стадии лимфомы проводят следующие исследования:
- Трепанобиопсия костного мозга.
- Методы лучевой диагностики — УЗИ, КТ, ПЭТ-КТ, МРТ.
- Также назначается ряд лабораторных анализов — развернутый анализ крови, определение маркеров парентеральных гепатитов, анализ на ВИЧ, биохимические исследования и др.
Лечение диффузной В-крупноклеточной лимфомы
Лечение диффузной лимфомы определяется исходя из следующих данных:
- Риски рецидива согласно IPI.
- Возраст пациента.
- Его состояние (сможет ли больной перенести высокодозную полихимиотерапию).
В качестве основных методов лечения крупноклеточной лимфомы применяется химиотерапия и в некоторых случаях облучение. Перед началом терапии фертильных пациентов обсуждается вопрос о криоконсервации гамет (половых клеток), поскольку лечение может вызвать бесплодие.

Главным критерием подбора терапии является международный прогностический индекс IPI, который включает следующие аспекты:
- Возраст. Младше 60 лет — 0 баллов, старше — 1 балл.
- Состояние пациента по ECOG (активность больного и способность к самообслуживанию). 1-2 балла по ECOG это 0 по IPI, и 3-4 балла по ECOG — это 1 балл по IPI.
- Уровень ЛДГ. В норме — 0 баллов по IPI, повышен — 1 балл.
- Стадия лимфомы. 1-2 — 0 баллов, 3-4 — 1 балл.
- Наличие более 1 экстранодальной зоны поражения. Нет — 0, да — 1.
Интерпретация IPI следующая:
- 0-1 балл — лимфома низкого риска.
- 2 балла — лимфома промежуточно низкого риска.
- 3 балла — лимфома промежуточно высокого риска.
- 4-5 баллов — лимфома высокого риска.
Пациенты из группы низкого и промежуточно-низкого риска начинают свое лечение с 6 циклов иммунохимиотерапии по протоколу R-CHOP-21. Эта схема, помимо цитостатиков, предполагает применение иммунотерапевтического препарата ритуксимаба. Такая схема позволяет добиться пятилетней выживаемости у 80% больных. Для пациентов с 3-4 стадией заболевания, лечение дополняется лучевой терапией на зоны массивного и экстранодального опухолевого поражения.

Лечение пациентов из групп промежуточно-высокого и высокого риска подбирается индивидуально с учетом возраста и состояния больного по шкале ECOG. Молодым пациентам с хорошим соматическим состоянием назначаются более агрессивные схемы лечения, предполагающие проведение высокодозной химиотерапии с трансплантацией гемопоэтических стволовых клеток (ГСК). При высоких рисках поражения нервной системы проводится несколько циклов интратекальной химиотерапии, когда препараты вводят в спинномозговой канал. Пожилым и слабым пациентам подбираются более щадящие схемы. Оценка эффективности лечения производится в середине циклов химиотерапии и после их окончания.
Восстановление после лечения крупноклеточной неходжкинской лимфомы
В целом, после того как у пациента диагностировали ремиссию, он должен регулярно наблюдаться у онколога. Сначала раз в три месяца, потом раз в полгода, а потом ежегодно. Если через пять лет после достижения ремиссии, у пациента нет признаков заболевания, его считают выздоровевшим и снимают с учета.
Рецидивы крупноклеточной лимфомы
При лечении диффузной В-крупноклеточной лимфомы долгосрочной ремиссии удается достичь в 70-80 % случаев, но у ряда пациентов возникает рецидив. Лечение рецидивов проводится по следующей схеме:
- Химиотерапия второй линии. Она должна подавить опухолевый рост, в то же время не нанося ущерба гемопоэтическим стволовым клеткам.
- Сбор ГСК.
- Высокодозная химиотерапия. Используются высокие дозы цитостатиков, которые уничтожают самые стойкие опухолевые клетки, но они же губят и кроветворение, поэтому чтобы его восстановить проводят следующий этап.
- Трансплантация гемопоэтических стволовых клеток, которая призвана восстановить кроветворение.
У пациентов из групп высокого риска, которые уже прошли аутологичную трансплантацию, и получили рецидив диффузной лимфомы, проводят аллогенную трансплантацию, т. е. используют стволовые клетки доноров. В этих случаях также есть шансы на полную ремиссию, но они не превышают 50%.
Осложнения диффузной В крупноклеточной лимфомы
Химиотерапия и лучевая терапия диффузной крупноклеточной лимфомы пагубно влияют не только на злокачественную опухоль, но и на все быстро делящиеся клетки. Это кроветворные клетки, эпителий кожи и слизистых оболочек и др. Поэтому в процессе лечения и восстановления большое внимание уделяется профилактике осложнений. В первую очередь, это борьба с инфекциями (бактериальными, вирусными, грибковыми), нарушением кровесвертывающей системы и работы пищеварительного тракта.
Помимо этого, в долгосрочной перспективе есть риск развития следующих осложнений:
- Возникновение других злокачественных опухолей: рак легкого, молочной железы, желудка, а также лейкозов и других видов лимфом. Наибольшие риски отмечаются в первые десятилетия после прекращения лечения.
- Кардиоваскулярные осложнения: нарушение работы миокарда, быстрый атеросклероз кровеносных сосудов, повреждение клапанов сердца и др.
- Поражение легких: пульмониты, пневмосклероз и пневмофиброз.
- Осложнение со стороны эндокринной системы: гипотиреоз, бесплодие, нарушение сперматогенеза.
Прогноз и профилактика крупноклеточной лимфомы
Прогноз течения диффузной В крупноклеточной лимфомы зависит от показателя IPI. Чем больше баллов, тем хуже прогноз. Для больных, имеющих 4-5 баллов (высокий риск), пятилетняя выживаемость колеблется в пределах 31%. При низких рисках пятилетняя выживаемость составляет около 91%.
Что касается специфической профилактики, то ее на сегодняшний день не существует, а все рекомендации для минимизации риска заболевания сводятся к ведению здорового образа жизни и правильному питанию.
Перспективные методы лечения диффузной В-крупноклеточной лимфомы
Каждый год только в США на диффузную В-крупноклеточную лимфому заболевает более 18,000 человек. Эта болезнь составляет 40% всех случаев неходжкинской лимфомы в мире. Риск заболеть повышается с возрастом, но в последнее десятилетие этот вид рака начал всё чаще встречаться у детей и подростков. О том, какие современные методы диффузной В-крупноклеточной лимфомы предлагают за рубежом, вы можете прочитать в этой статье.
Что такое диффузная В-крупноклеточная лимфома?

Диффузная В-крупноклеточная лимфома (ДККЛ) – один из самых распространенных видов рака крови. Она поражает клетки, которые обычно занимаются защитой организма от инфекций и чужеродных агентов. Основной симптом, на который обращают внимание пациенты – увеличение лимфоузлов.
Врачи могут обнаружить лимфому, посмотрев на кровь сквозь микроскоп. В-лимфоциты, пораженные раком – крупнее нормальных, и склонны держатся поодиночке.
В-крупноклеточная лимфома вызывается мутацией в иммунных клетках, а конкретно – на трёх отдельных участках ДНК. Чем больше таких мутаций, тем тяжелее этот вид рака лечится.
Средняя выживаемость при диффузной В-крупноклеточной лимфоме составляет 71%.
В зависимости от внешнего вида, поведения и типа пораженного белка, ДККЛ делится на 5 подтипов:
Обычная диффузная В-крупноклеточная лимфома;
Обычная диффузная В-крупноклеточная лимфома с поражением кожи;
Диффузная В-крупноклеточная лимфома с избытком Т-клеток и гистиоцитов;
Диффузная В-крупноклеточная лимфома с выявленным вирусом Эпштейна-Барра;
Неспецифическая диффузная В-крупноклеточная лимфома.
Диффузная В-крупноклеточная лимфома – агрессивное заболевание, что быстро поражает организм человека. Из-за этого она часто диагностируется на поздних стадиях.
Большинство пациентов, страдающих от этого вида рака – люди старше 60 лет. У мужчин он встречается немного чаще.
Среди факторов риска развития В-крупноклеточной лимфомы выделяют:
Аутоиммунные заболевания (ревматоидный артрит, волчанка);
Перенесенные длительные воспалительные процессы;
Перенесенная в прошлом лимфома;
ВИЧ;
Гепатит С;
Пересадка органов;
Наличие кровных родственников, что болели лимфомой;
Лишний вес в подростковом возрасте.
История Шерил Уильямс
Шерил Уильямс была обычной пенсионеркой из Чикаго, бывшей преподавательнецей детского сада. Жизнь Шерил резко изменилась в 2011 году, когда на приеме у врача она услышала диагноз – “диффузная В-крупноклеточная лимфома 4 стадии”.
Благодаря быстрой реакции врачей, а также с поддержкой семьи и друзей, Шерил удалось одолеть этот недуг. Она прошла курс химиотерапии, предписанный онкологом. Каждый раз, когда нужно было проходить сканирование или сдавать кровь на анализы, она боялась услышать наихудшие новости. Наконец наступил тот день, когда доктора сообщили Шерил о ремиссии. Её организм был свободен от рака.
После достижения ремиссии на протяжении следующих 5 лет пациентам нужно проходить регулярные проверки. Это необходимо для того, чтобы врачи могли вовремя обнаружить возвращение рака. После истечения пятилетного срока риск возникновения рецидива минимальный.
В случае Шерил, именно это и произошло. Перед третьей годовщиной своего выздоровления она узнала, что лимфома вернулась. Болезнь была обнаружена на рутинной проверке, после чего женщину попросили пройти УЗИ и биопсию груди. Анализы подтвердили, что это всё тот же вид рака.
У онкобольных с диффузной В-крупноклеточной лимфомой в стадии рецидива меньшие шансы на выздоровление. Этот тип рака хуже поддается химиотерапии.
К счастью для Шерил, и во второй раз она смогла победить рак. Врач прописал ей курс более сильной иммунотерапии, а также пересадку костного мозга. С тех пор прошло пять лет. Сегодня Шерил – счастливая, а самое главное – абсолютно здоровая женщина.
Читать: ТОП-5 клиник Европы по лечению рака
Как обычно лечат диффузную В-крупноклеточную лимфому?
Основной способ лечения диффузной В-крупноклеточной лимфомы – это химиотерапия. Смысл этого метода заключается в том, что больной принимает препараты с токсическим влиянием на злокачественные образования. Из-за этих лекарств раковые клетки перестают размножаться и умирают.
Химиотерапия – название большой группы препаратов. Для каждого вида рака есть различные курсы химиотерапии, между которыми врач выбирает самый оптимальный. Подбор конкретного протокола зависит от типа и стадии онкозаболевания, возраста больного, состояния его здоровья. Доктор подбирает комбинацию лекарств, которая давала бы максимальный результат при минимальных побочных эффектах.
Диффузная В-крупноклеточная лимфома ранней стадии лечится 3-6 циклами химиотерапии, что проводятся на протяжении интервалов в 21 день. Циклы приема лекарств чередуются с циклами отдыха, во время которых организм восстанавливает свои силы.
На 3-4 стадии больному может понадобиться более 6 циклов химиотерапии. Рецидивирующий рак тяжелее поддается лечению, поэтому врачи используют более агрессивные методы.
Более легкие формы химиотерапии принимаются только раз в неделю. Интенсивные курсы требуют амбулаторного пребывания больного на протяжении 5-7 дней. Лекарство поступает в организм постепенно, за счет чего ослабляется токсичный эффект.
Эффективность химиотерапии для лечения диффузной В-крупноклеточной лимфомы – 60-80%.
Современная химиотерапия за рубежом обладает меньшим количеством побочных эффектов и легче переносится пациентами.
Международный прогностический индекс
Международный прогностический индекс – система, позволяющая определить врачам уровень риска возникновения рецидива. Показатель формируется на основе:
Возраста больного;
Состояния его здоровья;
Стадии заболевания;
Уровень нормальной повседневной активности;
Наличие метастаз;
Количества лактатдегидрогеназы в крови.
Чем выше бал по прогностическому индексу, тем выше риск рецидива.
Пересадка костного мозга
Если больному не помогает химиотерапия, врачи прибегают к пересадке костного мозга.
Костный мозг – часть ткани трубчатых костей человека, что создает клетки крови, включая иммунные. Во время пересадки человеку вводят здоровые клетки вместо старых. Существует три типа процедуры:
аутологическая (с помощью собственных клеток организма);
аллогенная от родственного донора;
аллогенная из банка доноров.
У больных, страдающих от диффузной В-крупноклеточной лимфомы, костный мозг слишком поражен для аутологической пересадки. Им доступен только аллогенный вариант.
Пациенты из СНГ предпочитают проходить пересадку костного мозга в зарубежных клиниках. По сравнению с отечественными больницами, у них выше процент успешности.
Перед пересадкой для лечения лимфомы нужно уничтожить зараженные раком клетки родного костного мозга. Обычно это происходит с помощью интенсивного курса химиотерапии.
При выборе страны важно обратить внимание надоступные варианты процедуры. Аутологическая трансплантация возможна во всех клиниках, что занимаются трансплантацией костного мозга. Но не везде разрешены пересадки от родственников или есть доступ к международной базе доноров.
Вследствии пересадки костного мозга человек на некоторое время теряет иммунитет, поэтому на период восстановления очень важны стерильные условия пребывания в больнице.
Читать: Где делают пересадку костного мозга и сколько это стоит
CAR-T ТЕРАПИЯ
CAR-T терапия считается самой перспективной альтернативой пересадке костного мозга и наибольшим прорывом в онкологии за последние годы. За её изобретение в 2018 году Джеймсу Эллисону и Тасуку Хёндзе была вручена Нобелевская премия.
CAR-T метод – разновидность иммунотерапии. Его суть состоит в том, что из кровотока человека извлекаются иммунные клетки – Т-лимфоциты. В специализированной лаборатории их изменяют, обучая распознавать и уничтожать раковые клетки. Вновь попадая в организм, Т-лимфоциты начинают бороться с злокачественными образованиями.
Особенно эффективной CAR-T оказывается против лимфомы детей и подростоков.
Использование CAR-T терапии возвращает больным, которые до этого прожили бы не более 6-ти месяцев, нормальную продолжительность жизни.
В некоторых случаях CAR-T терапия способна вызывать побочные эффекты: синдром выброса цитокинов (симптомы похожи на простуду) и неврологическую токсичность. Тогда врач прописывает дополнительные лекарства.
CAR-T терапию используют, если обычные методы лечения оказались неэффективными, но она дороже пересадки костного мозга.
Привлекающие Т-клетки биспецифические активаторы (BiTE)
Еще одна разновидность иммунотерапии, эффективная при лечении диффузной В-крупноклеточной лимфомы – BiTE-терапия. Как и в предыдущем методе, суть состоит в манипуляциях над Т-лимфоцитами. Однако, вместо того, чтобы извлекать их из больного, изменения происходят в самом организме. Лекарство, что вводится в кровь, “склеивает” лимфоциты с раковыми опухолями.
Преимущество этого метода в том, что для его проведения не нужно доступа к высокотехнологичным лабораториям. Это делает лечение быстрее и дешевле.
Этот метод уже испытывали на группах больных с диффузной В-крупноклеточной лимфомой, которым не помогла химиотерапия высокими дозами и CAR-T терапия. BiTE-терапия сработала в 89% случаев.
Пока этот метод терапии находится на стадии исследований. Если он успешно пройдет все этапы и получит одобрение FDA, BiTE-терапия станет доступной в многих зарубежных клиниках.
Читать: CAR-T клеточная терапия – новый метод лечения рака
Где за рубежом лечат диффузную В-крупноклеточною лимфому?
Много зарубежных клиник, что специализируются на онкогематологии, могут похвастаться высоким процентом выживаемости своих пациентов. Страны, которые пользуются популярностью среди пациентов из СНГ – Германия, Израиль, Испания, Турция, Индия и Южная Корея.
Цена на лечение зависит от многих факторов, в первую очередь – стадии заболевания и необходимости пересадки костного мозга. Учитывается состояние больного, уровень больницы и лечащего врача, цена препаратов и т.д.
| Страна | Стоимость химиотерапии |
|---|---|
| Германия | от €2,500 за курс |
| Израиль | от $2,000 за курс |
| Испания | от €2,000 за курс |
| Турция | от $1,500 за курс |
| Индия | от $1,000 за курс |
| Южная Корея | от $2,000 за курс |
В Израиле хорошей репутацией пользуются доктора Оделия Гур и Левин Дрор, что практикуют в клинике Ихилов. Кроме того, врачи этой больницы имеют большой опыт в лечении детских онкологий.
В Испании лимфому следует лечить в центрах Киронсалюд, Текнон или в университетской клинике Наварры. Самый известный специалист страны – доктор Эмили Монтсеррат.
В Турции на лечении неходжкинских лимфом специализируется много больниц – Лив, Коч, Мемориал, Медикана, Медикал Парк. Одно из преимуществ лечения в Турции – цена ниже на 20-40% в сравнении с Израилем и Германией. При этом врачи используют такое же оборудование и протоколы, а больницы владеют сертификатами качества и признаны мировым медицинским сообществом.
Читать: Сколько стоит лечение лимфомы и где ее лечить
Резюме
Диффузная В-крупноклеточная лимфома – вид рака крови, которому характерно быстрое и агрессивное течение. Из-за этого её часто обнаруживают уже на поздних стадиях.
Традиционная форма лечения ДККЛ – химиотерапия. При лимфоме 4 стадии или рецидиве может использоваться пересадка костного мозга от донора.
Популярность обретают альтернативные методы лечения – иммунотерапия, в частности – CAR-T терапия. Чаще всего она используется в случае, если обычные методы лечения оказались бессильны. BiTE – еще один перспективный вариант иммунотерапии, что проходит тестирование.
Эффективность лечения диффузной В-крупноклеточной лимфомы современными методами может достигать 89%.
Если Вас интересует лечение диффузной В-крупноклеточной лимфомы или остались вопросы, оставьте свою заявку на сайте MediGlobus. Наши врачи-координаторы свяжутся с Вами и помогут с подбором клиники и организацией поездки.
Неходжкинские лимфомы ( Лимфосаркома )

Неходжкинские лимфомы – опухолевые заболевания лимфатической системы, представленные злокачественными B- и T-клеточными лимфомами. Первичный очаг может возникать в лимфатических узлах либо других органах и в дальнейшем метастазировать лимфогенным или гематогенным путем. Клиника лимфом характеризуется лимфаденопатией, симптомами поражения того или иного органа, лихорадочно-интоксикационным синдромом. Диагностика основывается на клинико-рентгенологических данных, результатах исследования гемограммы, биоптата лимфоузлов и костного мозга. Противоопухолевое лечение включает курсы полихимиотерапии и лучевой терапии.
МКБ-10



- Причины
- Патогенез
- Классификация
- Симптомы
- Осложнения
- Диагностика
- Лечение
- Прогноз и профилактика
- Цены на лечение
Общие сведения
Неходжкинские лимфомы (НХЛ, лимфосаркомы) – различные по морфологии, клиническим признакам и течению злокачественные лимфопролиферативные опухоли, отличные по своим характеристикам от лимфомы Ходжкина (лимфогранулематоза). В зависимости от места возникновения первичного очага гемобластозы делятся на лейкозы (опухолевые поражения костного мозга) и лимфомы (опухоли лимфоидной ткани с первичной внекостномозговой локализацией). На основании отличительных морфологических признаков лимфомы, в свою очередь, подразделяются на ходжкинские и неходжкинские; к числу последних в гематологии относят В- и Т-клеточные лимфомы. Неходжкинские лимфомы встречаются во всех возрастных группах, однако более половины случаев лимфосарком диагностируется у лиц старше 60 лет. Средний показатель заболеваемости среди мужчин составляет 2-7 случая, среди женщин – 1-5 случаев на 100 000 населения. В течение последних лет прослеживается тенденция к прогрессирующему увеличению заболеваемости.
Причины
Этиология лимфосарком достоверно неизвестна. Более того, причины лимфом различных гистологических типов и локализаций существенно варьируются. В настоящее время правильнее говорить о факторах риска, повышающих вероятность развития лимфомы, которые на данный момент хорошо изучены. Влияние одних этиофакторов выражено значительно, вклад других в этиологию лимфом весьма несущественен. К такого рода неблагоприятным предпосылкам относятся:
- Инфекции. Наибольшим цитопатогенным эффектом на лимфоидные клетки обладает вирус иммунодефицита человека (ВИЧ), гепатита С, Т-лимфотропный вирус 1 типа. Доказана связь инфицирования вирусом Эпштейна-Барр с развитием лимфомы Беркитта. Известно, что инфекция Helicobacter pylori, ассоциированная с язвенной болезнью желудка, может вызывать развитие лимфомы той же локализации.
- Дефекты иммунитета. Риск возникновения лимфом повышается при врожденных и приобретенных иммунодефицитах (СПИДе, синдроме Вискотта-Олдрича, Луи-Бар, Х-сцепленном лимфопролиферативном синдроме и др.). У пациентов, получающих иммуносупрессивную терапию по поводу трансплантации костного мозга или органов, вероятность развития НХЛ увеличивается в 30-50 раз.
- Сопутствующие заболевания. Повышенный риск заболеваемости НХЛ отмечается среди пациентов с ревматоидным артритом, красной волчанкой, что может быть объяснено как иммунными нарушениями, так и использованием иммуносупрессивных препаратов для лечения данных состояний. Лимфома щитовидной железы обычно развивается на фоне аутоиммунного тиреоидита.
- Токсическое воздействие. Прослеживается причинно-следственная связь между лимфосаркомами и предшествующим контактом с химическими канцерогенами (бензолом, инсектицидами, гербицидами), УФ-излучением, проведением лучевой терапии по поводу онкологического заболевания. Прямое цитопатическое действие оказывают цитостатические препараты, применяемые для химиотерапии.
Патогенез
Патологический лимфогенез инициируется тем или иным онкогенным событием, вызывающим нарушение нормального клеточного цикла. В этом могут быть задействованы два механизма – активация онкогенов либо подавление опухолевых супрессоров (антионкогенов). Опухолевый клон при НХЛ в 90% случаев формируется из В-лимфоцитов, крайне редко – из Т-лимфоцитов, NK- клеток или недифференцированных клеток.
Для различных типов лимфом характерны определенные хромосомные транслокации, которые приводят к подавлению апоптоза, утрате контроля над пролиферацией и дифференцировкой лимфоцитов на любом этапе. Это сопровождается появлением клона бластных клеток в лимфатических органах. Лимфоузлы (периферические, медиастинальные, мезентериальные и др.) увеличиваются в размерах и могут нарушать функцию близлежащих органов. При инфильтрации костного мозга развивается цитопения. Разрастание и метастазирование опухолевой массы сопровождается кахексией.
Классификация
Лимфосаркомы, первично развивающиеся в лимфоузлах называются нодальными, в других органах (небной и глоточных миндалинах, слюнных железах, желудке, селезенке, кишечнике, головном мозге, легких, коже, щитовидной железе и др.) – экстранодальными. По структуре опухолевой ткани НХЛ делятся на фолликулярные (нодулярные) и диффузные. По темпам прогрессирования лимфомы классифицируются на индолентные (с медленным, относительно благоприятным течением), агрессивные и высоко агрессивные (с бурным развитием и генерализацией). При отсутствии лечения больные с индолентными лимфомами живут в среднем 7 – 10 лет, с агрессивными – от нескольких месяцев до 1,5-2 лет.
Современная классификация насчитывает свыше 30 различных видов лимфосарком. Большая часть опухолей (85%) происходит из В-лимфоцитов (В-клеточные лимфомы), остальные из Т-лимфоцитов (Т-клеточные лимфомы). Внутри этих групп существуют различные подтипы неходжкинских лимфом. Группа В-клеточных опухолей включает:
- диффузную В-крупноклеточную лимфому – самый распространенный гистологический тип лимфосарком (31%). Характеризуется агрессивным ростом, несмотря на это почти в половине случаев поддается полному излечению.
- фолликулярную лимфому – ее частота составляет 22% от числа НХЛ. Течение индолентное, однако возможна трансформация в агрессивную диффузную лимфому. Прогноз 5-летней выживаемости – 60-70%.
- мелкоклеточную лимфоцитарную лимфомуи хронический лимфоцитарный лейкоз – близкие типы НХЛ, на долю которых приходится 7% от их числа. Течение медленное, но плохо поддающееся терапии. Прогноз вариабелен: в одних случаях лимфосаркома развивается в течение 10 лет, в других – на определенном этапе превращается в быстрорастущую лимфому.
- лимфому из мантийных клеток – в структуре НХЛ составляет 6%. Пятилетний рубеж выживаемости преодолевает лишь 20% больных.
- В-клеточные лимфомы из клеток маргинальной зоны – делятся на экстранодальные (могут развиваться в желудке, щитовидной, слюнных, молочных железах), нодальные (развиваются в лимфоузлах), селезеночную (с локализацией в селезенке). Отличаются медленным локальным ростом; на ранних стадиях хорошо поддаются излечению.
- В-клеточную медиастинальную лимфому – встречается редко (в 2% случаев), однако в отличие от других типов поражает преимущественно молодых женщин 30-40 лет. В связи с быстрым ростом вызывает компрессию органов средостения; излечивается в 50% случаев.
- макроглобулинемию Вальденстрема (лимфоплазмоцитарную лимфому) – диагностируется у 1% больных с НХЛ. Характеризуется гиперпродукцией IgM опухолевыми клетками, что приводит к повышению вязкости крови, сосудистым тромбозам, разрывам капилляров. Может иметь как относительно доброкачественное (с выживаемостью до 20 лет), так и скоротечное развитие (с гибелью пациента в течение 1-2 лет).
- волосатоклеточный лейкоз– очень редкий тип лимфомы, встречающийся у лиц пожилого возраста. Течение опухоли медленное, не всегда требующее лечения.
- лимфому Беркитта – на ее долю приходится около 2% НХЛ. В 90% случаев опухоль поражает молодых мужчин до 30 лет. Рост лимфомы Беркитта агрессивный; интенсивная химиотерапия позволяет добиться излечение половины больных.
- лимфому центральной нервной системы – первичное поражение ЦНС может затрагивать головной или спинной мозг. Чаще ассоциируется с ВИЧ-инфекцией. Пятилетняя выживаемость составляет 30%.
Неходжкинские лимфомы Т-клеточного происхождения представлены:
- Т-лимфобластной лимфомой или лейкозом из клеток-предшественников – встречается с частотой 2%. Различаются между собой количеством бластных клеток в костном мозге: при 25% – как лейкоз. Диагностируется преимущественно у молодых людей, средний возраст заболевших – 25 лет. Худший прогноз имеет Т-лимфобластный лейкоз, показатель излечения при котором не превышает 20%.
- периферическими Т-клеточными лимфомами, включающими кожную лимфому (синдром Сезари, грибовидный микоз), ангиоиммунобластную лимфому, экстранодальную лимфому из естественных киллеров, лимфому с энтеропатией, панникулитоподобную лимфому подкожной клетчатки, крупноклеточную анапластическую лимфому. Течение большей части Т-клеточных лимфом быстрое, а исход неблагоприятный.
Симптомы
Варианты клинических проявлений НХЛ сильно варьируются в зависимости от локализации первичного очага, распространенности опухолевого процесса, гистологического типа опухоли и пр. Все проявления лимфосарком укладываются в три синдрома: лимфаденопатии, лихорадки и интоксикации, экстранодального поражения. В большинстве случаев первым признаком НХЛ служит увеличение периферических лимфоузлов. Вначале они остаются эластичными и подвижными, позднее сливаются в обширные конгломераты. Одновременно могут поражаться лимфоузлы одной или многих областей. При образовании свищевых ходов необходимо исключить актиномикоз и туберкулез.
Такие неспецифические симптомы лимфосарком, как лихорадка без очевидных причин, ночная потливость, потеря веса, астения в большинстве случаев указывают на генерализованный характер заболевания. Среди экстранодальных поражений доминируют неходжкинские лимфомы кольца Пирогова-Вальдейера, ЖКТ, головного мозга, реже поражаются молочная железа, кости, паренхима легких и др. органы. Лимфома носоглотки при эндоскопическом исследовании имеет вид опухоли бледно-розового цвета с бугристыми контурами. Часто прорастает верхнечелюстную и решетчатую пазуху, орбиту, вызывая затруднение носового дыхания, ринофонию, снижение слуха, экзофтальм.
Первичная лимфосаркома яичка может иметь гладкую или бугристую поверхность, эластическую или каменистую плотность. В некоторых случаях развивается отек мошонки, изъязвление кожи над опухолью, увеличение пахово-подвздошных лимфоузлов. Лимфомы яичка предрасположены к ранней диссеминации с поражением второго яичка, ЦНС и др.
Лимфома молочной железы при пальпации определяется как четкий опухолевый узел или диффузное уплотнение груди; втяжение соска нехарактерно. При поражении желудка клиническая картина напоминает рак желудка, сопровождаясь болями, тошнотой, потерей аппетита, снижением веса. Абдоминальные лимфосаркомы могут проявлять себя частичной или полной кишечной непроходимостью, перитонитом, синдромом мальабсорбции, болями в животе, асцитом. Лимфома кожи проявляется зудом, узелками и уплотнением красновато-багрового цвета. Первичное поражение ЦНС более характерно для больных СПИДом – течение лимфомы данной локализации сопровождаются очаговой или менингеальной симптоматикой.
Осложнения
Наличие значительной опухолевой массы может вызывать сдавление органов с развитием жизнеугрожащих состояний. При поражении медиастинальных лимфоузлов развивается компрессия пищевода и трахеи, синдром сдавления ВПВ. Увеличенные внутрибрюшные и забрюшинные лимфатические узлы могут вызвать явления кишечной непроходимости, лимфостаза в нижней половине туловища, механической желтухи, компрессии мочеточника. Прорастание стенок желудка или кишечника опасно возникновением кровотечения (в случае аррозии сосудов) или перитонита (при выходе содержимого в брюшную полость). Иммуносупрессия обусловливает подверженность пациентов инфекционным заболеваниям, представляющим угрозу для жизни. Для лимфом высокой степени злокачественности характерно раннее лимфогенное и гематогенное метастазирование в головной и спинной мозг, печень, кости.
Диагностика
Вопросы диагностики неходжкинских лимфом находятся в компетенции онкогематологов. Клиническими критериями лимфосаркомы служат увеличение одной или нескольких групп лимфоузлов, явления интоксикации, экстранодальные поражения. Для подтверждения предполагаемого диагноза необходимо проведение морфологической верификации опухоли и инструментальной диагностики:
- Исследование клеточного субстрата опухоли. Выполняются диагностические операции: пункционная или эксцизионная биопсия лимфоузлов, лапароскопия, торакоскопия, аспирационная пункция костного мозга с последующими иммуногистохимическими, цитологическими, цитогенетическими и другими исследованиями диагностического материала. Кроме диагностики, установление структуры НХЛ важно для выбора тактики лечения и определения прогноза.
- Методы визуализации. Увеличение медиастинальных и внутрибрюшных лимфоузлов обнаруживается с помощью УЗИ средостения, рентгенографии и КТ грудной клетки, брюшной полости. В алгоритм обследования по показаниям входят УЗИ лимфатических узлов, печени, селезенки, молочных желез, щитовидной железы, органов мошонки, гастроскопия. С целью стадирования опухоли проводится МРТ внутренних органов; в выявлении метастазов информативны лимфосцинтиграфия, сцинтиграфия костей.
- Лабораторная диагностика. Направлена на оценку факторов риска и функции внутренних органов при лимфомах различных локализаций. В группе риска производится определение ВИЧ-антигена, анти-HCV. Изменение периферической крови (лимфоцитоз) характерно для лейкемизации. Во всех случаях исследуется биохимический комплекс, включающий печеночные ферменты, ЛДГ, мочевую кислоту, креатинин и др. показатели. Своеобразным онкомаркером НХЛ может служить b2-микроглобулин.
Дифференцировать неходжкинские лимфомы приходится с лимфогранулематозом, метастатическим раком, лимфаденитами, возникающими при туляремии, бруцеллезе, сифилисе, туберкулезе, токсоплазмозе, инфекционном мононуклеозе, гриппе, СКВ и др. При лимфомах конкретных локализаций проводятся консультации профильных специалистов: оториноларинголога, гастроэнтеролога, маммолога и т.д.
Лечение
Варианты лечения неходжкинских лимфом включают оперативный метод, лучевую терапию и химиотерапию. Выбор методики определяется морфологическим типом, распространенностью, локализацией опухоли, сохранностью и возрастом больного. В современной онкогематологии приняты протоколы лечения лимфосарком, базирующиеся на использовании:
- Химиотерапии. Наиболее часто лечение лимфом начинают с курса полихимиотерапии. Этот метод может являться самостоятельным или сочетаться с лучевой терапией. Комбинированная химиолучевая терапия позволяет достичь более длительных ремиссий. Лечение продолжается до достижения полной ремиссии, после чего необходимо проведение еще 2-3 консолидирующих курсов. Возможно включение в циклы лечения гормонотерапии.
- Хирургических вмешательств. Обычно применяется при изолированном поражении какого-либо органа, чаще – ЖКТ. По возможности операции носят радикальный характер – выполняются расширенные и комбинированные резекции. В запущенных случаях, при угрозе перфорации полых органов, кровотечения, непроходимости кишечника могут выполняться циторедуктивные вмешательства. Хирургическое лечение обязательно дополняется химиотерапией.
- Лучевой терапии. В качестве монотерапии лимфом применяется только при локализованных формах и низкой степени злокачественности опухоли. Кроме этого, облучение может быть использовано и в качестве паллиативного метода при невозможности проведения других вариантов лечения.
- Дополнительных схем лечения. Из альтернативных методов хорошо себя зарекомендовала иммунохимиотерапия с применением интерферона, моноклональных антител. С целью консолидации ремиссии применяется трансплантация аутологичного или аллогенного костного мозга и введение периферических стволовых клеток.
Прогноз и профилактика
Прогноз при неходжкинских лимфомах различен, зависит, главным образом, от гистологического типа опухоли и стадии выявления. При местнораспространенных формах долгосрочная выживаемость в среднем составляет 50-60%, при генерализованных – всего 10-15%. Неблагоприятными прогностическими факторами служат возраст старше 60 лет, III-IV стадии онкопроцесса, вовлечение костного мозга, наличие нескольких экстранодальных очагов. Вместе с тем, современные протоколы ПХТ во многих случаях позволяют добиться долгосрочной ремиссии. Профилактика лимфом коррелирует с известными причинами: рекомендуется избегать инфицирования цитопатогенными вирусами, токсических воздействий, чрезмерной инсоляции. При наличии факторов риска необходимо проходить регулярное обследование.
В клеточная лимфома крупноклеточная, диффузная: шансы на выживание, прогноз
Диффузная В-клеточная крупноклеточная лимфома относится к агрессивному классу лимфом.
«В-клеточная», потому что состоит из заболевших В-клеток, а В-клетки (В-лимфоциты) – это клетки-следователи, они ищут и передают сообщения о клетках-чужаках другим членам иммунитета. Опорными пунктами иммунной системы являются лимфатические узлы, которые имеют свое строение и разделены на области. Каждая область лимфатического узла выполняет определенную работу. При диффузной В-крупноклеточной лимфоме строение лимфатического узла нарушено, опухолевые клетки находятся во всех областях, то есть диффузно, поэтому и лимфома называется «диффузной».
Одиночные лимфомные опухоли могут появляться в любой точке тела, хотя чаще при этой болезни увеличиваются лимфатические узлы группами на шее, подмышками, в паху и в других местах. Увеличенные узлы можно прощупать руками или найти с помощью рентгена и ультразвука (УЗИ). У больного могут появляться такие признаки болезни, которые называются «В-симптомы»: он быстро худеет, у него поднимается температура и появляется сильная потливость вечером и ночью. Диффузная В-клеточная крупноклеточная лимфома может возникнуть сама или ее могут вызвать другие лимфомы, которые развиваются в организме больного медленно, так называемые индолентные, вялотекущие лимфомы.
Существуют разные виды диффузной В-крупноклеточной лимфомы:
- обычная диффузная В-крупноклеточная лимфома (самая частая),
- диффузная В-крупноклеточная лимфома с избытком Т-клеток и гистиоцитов,
- диффузная В-крупноклеточная лимфома с первичным поражением головного мозга,
- первично-кожная диффузная В-крупноклеточная лимфома с поражением ног,
- диффузная В-крупноклеточная лимфома с выявленным вирусом Эпштейна-Барра,
- диффузная В-крупноклеточная лимфома на фоне хронического воспаления.
Диффузная В-клеточная крупноклеточная лимфома быстро нарастает, прогрессирует, но в целом хорошо поддается лечению.
Диагноз
Чтобы поставить правильный диагноз, обязательно берут кусочек опухоли или увеличенного лимфатического узла для морфологического и иммуногистохимического исследования. Только после такого сложного комплексного анализа можно быть уверенным в диагнозе и провести эффективное лечение.
Для того чтобы точно узнать, есть ли увеличенные лимфатические узлы и каково их количество в тех частях тела, которые нельзя увидеть при внешнем осмотре, необходимо сделать компьютерную томографию грудной клетки, брюшной полости и малого таза. Стадию болезни устанавливают по системе «Ann-Arbor», которая показывает, какие органы и лимфатические узлы затронуты болезнью.
Для того чтобы точно узнать, есть ли увеличенные лимфатические узлы и каково их количество в тех частях тела, которые нельзя увидеть при внешнем осмотре, необходимо сделать компьютерную томографию грудной клетки, брюшной полости и малого таза. Стадию болезни устанавливают по системе «Ann-Arbor», которая показывает, какие органы и лимфатические узлы затронуты болезнью.
Лечение
Обычную диффузную В-крупноклеточную лимфому чаще всего начинают лечить по общепринятой схеме «СНОР», с добавлением лекарства «ритуксимаб». Как правило, проводят 6 курсов, но возможны варианты как по длительности, так и по выбору схемы лечения в зависимости от состояния здоровья пациента и особенностей болезни. Более редкие виды диффузной В-крупноклеточой лимфомы имеют свои особенности лечения. Между курсами, для оценки их эффективности, может потребоваться контрольная компьютерная томография. Если первое лечение не помогает, то терапией «второй линии» является высокодозная химиотерапия с поддержкой собственными стволовыми клетками.
Применение современных схем лечения позволяет полностью излечить около 75% пациентов.
Диффузная В-крупноклеточная лимфома у взрослых. Клинические рекомендации.
Диффузная В-крупноклеточная лимфома у взрослых
- Национальное гематологическое общество Российское профессиональное общество онкогематологов
Оглавление
- Ключевые слова
- Список сокращений
- Термины и определения
- 1. Краткая информация
- 2. Диагностика
- 3. Лечение
- 4. Реабилитация
- 5. Профилактика и диспансерное наблюдение
- 6. Дополнительная информация, влияющая на течение и исход заболевания
- Критерии оценки качества медицинской помощи
- Список литературы
- Приложение А1. Состав рабочей группы
- Приложение А2. Методология разработки клинических рекомендаций
- Приложение Б. Алгоритмы ведения пациента
- Приложение В. Информация для пациентов
Ключевые слова
клинические рекомендации 2016 г
Терапия 1-й линии
Терапия 2-й линии
Список сокращений
ДВКЛ – диффузная В-крупноклеточная лимфома
КТ – компьютерная томография
МРТ – магнитно-резонансная томография
УЗИ – ультразвуковое исследование
ЛТ – лучевая терапия
ВДХТ с аутоТГСК – высокодозная химиотерапия с последующей трасплантацией аутологичных гемопоэтических стволовых клеток.
ПМВКЛ – первичная медиастинальная В-крупноклеточная лимфома
ЦНС – центральная нервная система
ПЭТ – позитронно-эмиссионная томография
Термины и определения
Первичная медиастинальная В-крупноклеточная лимфома (ПМВКЛ) относится к первичным экстранодальным опухолям и происходит из В-клеток мозгового слоя вилочковой железы.
Диффузная В-крупноклеточная лимфома (ДВКЛ) является гетерогенной группой лимфатических опухолей с различными клиническими, морфологическими, иммунофенотипическими, цитогенетическими проявлениями и с разным ответом на терапию. Субстратом опухоли являются крупные В-лимфоидные клетки с выраженным атипизмом и полиморфизмом, с размером ядра в два и более раз превышающим размер ядра малого лимфоцита. Опухолевые клетки в большинстве случаев располагаются диффузно, но могут быть и разбросанными среди зрелых В-лимфоцитов, иногда на фоне Т-клеточного окружения, или формировать очаговые скопления [1-3].
Нодальная ДВКЛ – заболевание с первичным и преимущественным поражением лимфоузлов
Экстранодальная ДВКЛ – заболевание с первичным поражением любого органа, кроме лимфоузлов
1. Краткая информация
1.1 Определение
ДВКЛ является гетерогенной группой лимфатических опухолей с различными клиническими, морфологическими, иммунофенотипическими, цитогенетическими проявлениями и с разным ответом на терапию. Субстратом опухоли являются крупные В-лимфоидные клетки с выраженным атипизмом и полиморфизмом, с размером ядра в два и более раз превышающим размер ядра малого лимфоцита. Опухолевые клетки в большинстве случаев располагаются диффузно, но могут быть и разбросанными среди зрелых В-лимфоцитов, иногда на фоне Т-клеточного окружения, или формировать очаговые скопления.
1.2 Этиология и патогенез.
В-клетки образуются в костном мозге, где происходит первичная перестройка генов, кодирующих синтез иммуноглобулинов. Гены вариабельного региона легких цепей (k или ?) собраны с помощью соединяющих (J – joing), а гены вариабельного региона тяжелых цепей – с помощью соединяющих (J) и разнообразных (D – diversity) сегментов.
Клетки с «успешной перестройкой» генов иммуноглобулинов (наивные В-клетки), покидают к/м и попадают во вторичные лимфоидные органы – л/у, миндалины, селезенку, пейеровы бляшки. В них В-клетки, подвергающиеся действию антигена, образуют фолликулы вместе с фолликулярными дендритными клетками и Т-клетками. В результате этого процесса формируются зародышевые центры во вторичных лимфоидных фолликулах. В зародышевом центре вторичных фолликулов наивные B-клетки, не имеющие адекватного антигена и не способные произвести функциональное антитело, подвергаются апоптозу, т.е. погибают. Но в В-клетках с адекватными антигенами и способными произвести функциональное антитело в зародышевом центре происходит переключение класса иммуноглобулина (IgM, IgD на IgG, IgA или IgE), а также соматическая гипермутация (замена одного нуклеотида в гипервариабельных регионах иммуноглобулинов), после чего В-клетки покидают фолликул, становясь окончательно дифференцированными плазматическими клетками или долгоживущими В-клетками памяти. Случайные неудачи в управлении этими процессами и играют решающую роль в развитии B-клеточных опухолей, в том числе ДВКЛ.
Патогенез ДВКЛ плохо изучен. Разнообразие клинических и морфологических проявлений ДВКЛ, иммунофенотипа позволяет предположить, что ДВКЛ является не единой нозологической формой, а группой лимфатических опухолей, имеющих близкий, но неодинаковый патогенез.
Это доказывают и молекулярно-цитогенетические исследования последних лет, позволившие выделить несколько вариантов ДВКЛ в зависимости от уровня дифференцировки опухолевых клеток, типов нарушения клеточных процессов, хромосомных аномалий [4].
В патогенезе ДВКЛ, вероятно, имеют значения многие гены, регулирующие события в зародышевых центрах, но изученным механизмом является перестройка гена Bcl-6, которая вызвана неправильным переключением класса иммуноглобулинов В-клеток в зародышевом центре. Ген Bcl-6 расположен в локусе 3q27 и экспрессируется исключительно В-клетками зародышевого центра.
В физиологических условиях ген Bcl-6 связывается с определенными регулирующими последовательностями ДНК, влияет на транскрипцию других генов, участвующих в В-клеточной активации и терминальной дифференцировке лимфоцитов. При перестройке локуса 3q27 происходит блок дальнейшей дифференцировки В-клеток в плазматические клетки, что приводит к бесконтрольной пролиферации В-клеток зародышевого центра.
1.3 Эпидемиология.
Диффузная В-крупноклеточная лимфома относится к наиболее распространенным вариантам лимфопролиферативных заболеваний взрослых (30-50% от всех неходжкинских лимфом). В возрасте до 18 лет частота этого варианта агрессивной В-клеточной опухоли не превышает 8-10%. Заболеваемость ДВКЛ составляет в среднем 4-5 на 100 000 населения в год. Риск развития опухоли увеличивается с возрастом и значительно выше у людей с серопозитивностью на вирус гепатита С, при наличии вируса иммунодефицита человека (ВИЧ), при аутоиммунных заболеваниях. Мужчины и женщины болеют ДВКЛ с приблизительно равной частотой [2].
1.4 Кодирование по МКБ 10 :
C83.3 Лимфома крупноклеточная (диффузная) – ретикулосаркома
ТИМОМЕГАЛИЯ И СИНДРОМ ПЛАТТЕРА
История относительно интенсивного изучения вилочковой железы ( glandula thymus, thymus ) насчитывает около 400 лет. Начало его относится к 1614 г., когда профессор Базельского университета Феликс Платтер описал происшедшую у него
История относительно интенсивного изучения вилочковой железы (glandula thymus, thymus) насчитывает около 400 лет. Начало его относится к 1614 г., когда профессор Базельского университета Феликс Платтер описал происшедшую у него на глазах внезапную смерть 5-месячного ребенка, при вскрытии трупа которого не было найдено никаких причин, способных объяснить смерть, кроме очень большого тимуса. Смерть этого ребенка тогда впервые связали с огромным тимусом, назвав данное состояние тимическим удушьем — asthma thymicum.
В середине XIX в. профессор Венского университета Карл Рокитанский высказал предположение об инкреторной функции вилочковой железы. Данная концепция была положена в основу представлений о тимико-лимфатическом статусе [1, 2] как врожденной особенности конституции, предрасполагающей к внезапной смерти от незначительных причин. В симптомокомплекс тимико-лимфатического статуса включались первичная гиперфункция вилочковой железы, гипоплазия надпочечников, сердца и аорты [1–5]. С этого времени причиной тимической смерти стали считать острую надпочечниковую недостаточность [3, 4]. Однако отсутствие характерных для тимико-лимфатического состояния признаков во многих случаях при такой смерти, в том числе и симптомов надпочечниковой недостаточности, позволило поставить под сомнение и данную концепцию.
В начале 1960-х гг. было доказано, что тимус является центральным органом иммунной системы. С этого времени вилочковая железа рассматривается в качестве железы внутренней секреции, действие гормонов которой направлено на дифференцировку тимических лимфоцитов. Открытие основной функции вилочковой железы — продукции Т-лимфоцитов и регуляции иммунитета, а также принадлежность ее к системе эндокринных желез позволяют рассматривать данный орган как коммутатор иммунной и эндокринной систем. В серии работ отечественных исследователей последних 20 лет доказывается, что дети с большой вилочковой железой относятся к числу иммунодефицитных лиц.
В 1970 г. проф. Т. Е. Ивановской [6] для обозначения увеличенной вилочковой железы вместо вышеуказанных терминов был предложен термин «тимомегалия». Под тимомегалией российские морфологи понимают увеличение объема и массы тимуса выше предельных возрастных значений с сохранением нормальной гистоархитектоники органа. Начиная с 1970 г., этот термин стал широко использоваться как морфологами, так и клиницистами.
Однако проведенные исследования показали, что тимомегалия является лишь одним из симптомов того состояния, которое существует у детей, имеющих увеличенную вилочковую железу [7]. В связи с отсутствием другого термина, отражающего сущность состояния, которое имеет место у детей с увеличенной вилочковой железой (соответствующей тимомегалии в понимании патологоанатомов), предлагаем называть его синдромом Платтера.
Термином «синдром Платтера» следует обозначать состояние, при котором клинические и параклинические методы обследования позволяют выявить у детей и подростков ряд специфических изменений организма, сочетающихся с первичной длительно сохраняющейся тимомегалией. Клиническое понимание тимомегалии при синдроме Платтера отражает морфологическое состояние вилочковой железы, при котором масса и объем тимуса превышают предельные возрастные значения при сохранении нормальной гистоархитектоники органа.
Диагностика тимомегалии в клинической практике
Ориентировочная диагностика тимомегалии может быть проведена с помощью объективного исследования: осмотра, пальпации, перкуссии. Однако оценка результатов во многом субъективна и зависит от опыта исследователя [7].
Из инструментальных методов, позволяющих объективно выявить тимомегалию, используются рентгенодиагностика и ультразвуковое исследование (УЗИ).
Рентгенодиагностика. Методологической основой рентгенодиагностики увеличенного тимуса является следующее положение: в норме у детей любого возраста на стандартных рентгенограммах грудной клетки в прямой проекции тень тимуса не должна выходить за пределы тени сосудистого пучка и сердца.
В целях диагностики тимомегалии на рентгенограммах грудной клетки в прямой проекции мы применяли кардио-тимико-торакальный индекс (КТТИ) по J. Gewolb [8] и вазокардиальный индекс (ВКИ) [7]. Оба индекса определяются на стандартных рентгенограммах грудной клетки в прямой проекции. КТТИ представляет собой частное от деления ширины сосудистого пучка на уровне бифуркации трахеи (точка carina) на ширину грудной клетки на уровне купола диафрагмы. ВКИ представляет собой частное от деления ширины сосудистого пучка на уровне бифуркации трахеи (точка carina) на максимальную ширину тени сердца. Максимальная ширина тени сердца определяется как сумма двух перпендикулярных отрезков, опущенных на вертикальную линию (проведенную через середину позвоночного столба) из максимально удаленных от нее точек сердца слева и справа (рис. 1).
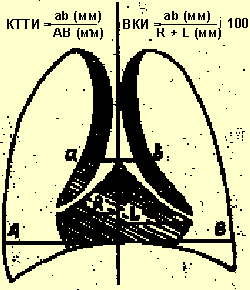 |
| Рисунок 1. Схема вычисления индексов КТТИ и ВКИ |
Выявление в средостении характерной овальной, треугольной или лентовидной тени с прямым, волнообразным или неровным контуром, расположенной с одной или обеих сторон сосудистого пучка, искажающей линию «сердечной талии» слева и/или вазокардиальный угол справа, позволяет сделать заключение о существовании у пациента тимомегалии. Наличие увеличенных значений КТТИ и ВКИ подтверждает данный диагноз. Рентгенологический метод, однако, информативен только для детей грудного и раннего возраста, поскольку у детей в возрасте старше 3 лет, как правило, тимус располагается за тенью сосудистого пучка и сердца. В таблице 1 приведены значения КТТИ и ВКИ у детей первых 3 лет жизни при разных степенях увеличения тимуса.
УЗИ. УЗИ тимуса в нашей стране впервые было проведено у новорожденных детей С. М. Воеводиным [9]. Путем клинико-морфологических сопоставлений нами были разработаны коэффициенты пересчета линейных параметров тимуса в более привычные для клиницистов формы (объема и массы органа). Округленные величины указанных коэффициентов, по нашим данным, равны 0,7 (для массы) и 0,5 (для объема). Таким образом, величины массы и объема тимуса при ультразвуковом сканировании вычисляются следующим образом:
М = 0,7 в a в b в c;
V = 0,5 в a в b в c,
где a, b, c — линейные параметры тимуса (ширина, длина, передне-задний размер, выраженные в см); 0,7 и 0,5 коэффициенты пересчета линейных параметров на массу и объем органа соответственно [10]. Для практических целей массу и объем вилочковой железы достаточно определять так, как это указано выше: без вычисления объема и массы отдельных долей.
Методом центильного распределения нами были определены нормальные значения массы и объема тимуса для детей в возрасте 1–24 мес (табл. 2). При превышении массы и объема тимуса выше верхних предельных значений нормы состояние ребенка следует расценивать как тимомегалию.
На основании анализа более 5000 рентгенограмм, выполненных у детей грудного возраста при подозрении на пневмонию, диагноз которой рентгенологически не был подтвержден, тимомегалия нами была выявлена у 35% пациентов 1 — 3 мес, у 15% детей в возрасте 3–6 мес, у 8% — в возрасте 7–15 мес, у 2% — в возрасте 16 — 36 мес, а в популяции детей старше 3 лет частота выявления тимомегалии не превышала 0,5% [8].
Наблюдение за одними и теми же детьми на протяжении длительного отрезка времени позволило нам сделать вывод, что тимомегалия в детском возрасте может быть как транзиторной, так и стойкой.
В 1993 г. нами совместно с О. В. Зайратьянцем была предложена классификация тимомегалии в детском возрасте на основе клинико-морфологических данных. Несколько измененный вариант указанной классификации приведен в табл. 3 (см. рубрику «Под стекло»). Наиболее частым вариантом тимомегалии в детском возрасте является синдром Платтера.
Анализ родословных позволил выявить у родственников первой и второй линии родства детей с синдромом Платтера высокую частоту встречаемости злокачественных новообразований, аутоиммунных заболеваний и заболеваний системы эндокринных органов. Так, злокачественные новообразования встречаются в каждой третьей семье, где есть дети с синдромом Платтера, с такой же частотой в этих семьях регистрируются эндокринные заболевания (преимущественно сахарный диабет и заболевания щитовидной железы). В семьях детей с синдромом Платтера статистически чаще, чем в семьях детей из выборки общей популяции, распространены аутоиммунные заболевания (диффузные болезни соединительной ткани) и туберкулез. Помимо этого, многие родители детей с указанным синдромом в дошкольном и младшем школьном возрасте относились к группе часто болеющих, т. е. к группе детей имевших повышенную склонность к острым респираторным вирусным инфекциям (ОРВИ).
Характер перечисленных заболеваний (склонность к вирусным, аутоиммунным, онкологическим заболеваниям, туберкулезу) может указывать на существование у близких родственников детей с синдромом Платтера недостаточной активности клеточного звена иммунной системы, поскольку, согласно современным воззрениям, все перечисленные заболевания находятся под контролем Т-клеточного иммунитета, формируемого вилочковой железой. При этом не совсем ясно, чем объясняется высокая отягощенность родственников детей с синдромом Платтера болезнями системы эндокринных органов. Однако не следует забывать о том, что эндокринные заболевания (в том числе сахарный диабет, диффузный токсический зоб, гипотиреоз) могут иметь аутоиммунный генез.
В таком случае у близких родственников детей с синдромом Платтера отчетливо просматривается тенденция к заболеваниям, связанным с иммунными механизмами клеточного типа, контролирующимися вилочковой железой. Это позволяет предполагать возможность передачи детям, у которых развился синдром Платтера, какого-то дефекта Т-звена иммунной системы.
Антенатальный период у большинства детей с синдромом Платтера осложняется хронической гипоксией, обусловленной острыми инфекционными заболеваниями или обострением хронических экстрагенитальных заболеваний матерей, наличием у них сосудистой дистонии, анемии, гестозов.
Осложненное течение интранатального периода отмечается не менее чем у 2/3 детей с синдромом Платтера. Наиболее частыми осложнениями этого периода явились стремительные или затяжные роды, приведшие к наложению щипцов или экстренному кесареву сечению.
Группа детей с указанным синдромом по фенотипу гетерогенна. 90% детей имеют мягкие округлые формы тела, некоторую пастозность тканей, относительно крупные черты уплощенного лица, прямой тип лба, широкоовальные глаза, относительно короткий нос с низким переносьем, средней величины или толстые губы, слабый изгиб профиля, короткую шею. Дети этой группы имеют как бы увеличенные поперечные размеры тела: широкое лицо, широкая грудная клетка, широкие плечи, широкие ладони и стопы, относительно короткие и широкие пальцы. Около 3/4 детей-европеоидов этой подгруппы имеют нежную, но несколько утолщенную, слабопигментированную белую кожу, для которой характерно быстрое развитие ожога после инсоляции, светлую окраску волос и глаз, слабое развитие мускулатуры, сниженную физическую активность. Дети данной подгруппы отличаются хорошим (и даже избыточным) аппетитом и высокими темпами роста.
В дошкольном и младшем школьном возрасте у них отмечаются хорошее развитие подкожного жирового слоя, слабое развитие мускулатуры, снижение общего тонуса и тургора тканей, а также физической активности, замедленная реакция на внешние раздражители, замедленное образование новых условных рефлексов, ослабление процессов внутреннего торможения с преобладанием реакций внешнего торможения.
Клиническая характеристика детей с синдромом Платтера включает характеристику системы лимфоидных органов, которая, согласно мнению большинства исследователей, отличается генерализованной гиперплазией системы лимфоидных органов: лимфатических узлов, лимфатических фолликулов корня языка, задней поверхности глотки, поверхности надгортанника, гипертрофией небных и глоточной миндалин. Наши наблюдения, однако, показали, что состояние периферических лимфоидных органов зависит от воздействия антигенного стимула — при отсутствии такого воздействия генерализованная гиперплазия периферического лимфоидного аппарата не развивается [7].
К числу особенностей физического развития детей грудного и раннего возраста относятся: высокие темпы увеличения длины тела, слабое развитие скелетной мускулатуры, несвоевременность и неправильный порядок прорезывания молочных зубов, относительно позднее начало самостоятельной ходьбы.
В эмоциональной сфере в первые 2–3 мес жизни обращает внимание менее выраженный комплекс оживления (за счет снижения двигательной активности).
Начало становления речи у детей с синдромом Платтера не отличалось от такового у детей соответствующего возраста в популяции в целом: гуление, слоги и первые слова у них появлялись своевременно.
К возрасту 10–11 мес они начинали произносить первые одно- и двусложные слова, к 1 году количество активно произносимых слов достигало 10–12. В дальнейшем 8% детей данной группы до 2-летнего возраста продолжали пользоваться только этими 10–12 словами. В возрасте 4,5–6 лет 25% детей этой группы страдали дислалией, требующей проведения корригирующих логопедических занятий в условиях детской поликлиники или в условиях специализированного детского сада. У отдельных пациентов нарушение, связанное с произношением звуков, сохранялось и в подростковом возрасте.
Состояние здоровья детей с синдромом Платтера. Здесь рассматриваются только те заболевания и патологические состояния, которые достоверно чаще встречались у детей с указанным синдромом, по сравнению с детьми из общей популяции. При анализе структуры выявленных заболеваний наибольшую по численности группу составили аномалии развития (выявлены у 80% наблюдавшихся пациентов) — пороки развития, множественные стигмы дизэмбриогенеза, биохимические дефекты. Пороки развития чаще проявлялись гипоплазией органов и тканей: широкие отверстия паховых каналов и пупочного кольца, септальные дефекты в сердце, гипоплазия аорты, сердца, легких, почек, щитовидной железы и др.
Вторая группа заболеваний была представлена патологическими состояниями нервной системы. Наряду с заболеваниями нервной системы, обусловленными биохимическими дефектами (синдром Менкеса; ганглиозидоз Gm1; фенилкетонурия; галактоземия), обращали на себя внимание такие синдромы, как гипертензионно-гидроцефальный (у 49%), фебрильных судорог (у 20%), мышечной гипотонии (у 18%) и вегетативно-висцеральной дисфункции (у 12,5%). Включение в комплекс обследования электрофизиологических методов, компьютерной и ядерно-магнитно-резонансной томографии привело к существенному увеличению частоты выявления гипертензионно-гидроцефального синдрома (выявлен у 90%). Указанный синдром у 2/3 детей сочетался с синдромами фебрильных судорог и вегетативно-висцеральных дисфункций, однако последние два синдрома у части детей встречались и изолированно.
Изолированный синдром вегетативно-висцеральных дисфункций характеризовался крайне выраженным полиморфизмом проявлений: нарушение терморегуляции, дыхания, сердечной деятельности, функции желудочно-кишечного тракта, пароксизмальное повышение артериального давления и другие расстройства. Мы склонны считать, что синдром вегето-висцеральных дисфункций у детей с синдромом Платтера встречается значительно чаще, чем у указанных 12,5% пациентов. На существование гиподиагностики синдрома вегето-висцеральных дисфункций могут указывать выявленные нами фенотипические особенности детей, в том числе очень хороший (скорее избыточный) аппетит, склонность к задержке жидкости, как бы немотивированный субфебрилитет. Наши наблюдения показали, что у большинства детей с якобы немотивированным субфебрилитетом последний хорошо контролировался мочегонными средствами, что явно указывает на связь этого симптома с ликворной гипертензией, в том числе с повышением давления в области третьего желудочка мозга — месте локализации центра терморегуляции.
У детей с синдромом Платтера обращал на себя внимание и синдром мышечной гипотонии, который у ряда пациентов был настолько сильно выражен, что специалисты высказывали предположение о наличии у них болезни Верднига–Гоффманна или миелодисплазии.
Таким образом, частое выявление у детей с синдромом Платтера внутренней гидроцефалии, изолированного синдрома вегето-висцеральных дисфункций, наличие вегетативных нарушений указывает на возможность нарушения функции диэнцефальной области. Изменения со стороны скелетной мускулатуры трактовать сложнее. Однако если учесть, что средний мозг, примыкающий к области сильвиева водопровода, контролирует регуляцию мышечного тонуса и сигналы к мышцам поступают через ретикулярную формацию по двум ретикулоспинальным путям [11], то это нарушение может быть рассмотрено с позиции существования у данного контингента детей внутренней гидроцефалии и расстройства ликвородинамики, поскольку гипоталамус, таламус и ретикулярная формация являются звеньями единой суперфункциональной системы, корригирующей гомеостатические программы наиболее сложных вегетативных, эндокринных, трофических и психических функций [12].
У 9% пациентов школьного возраста с синдромом Платтера зарегистрированы заболевания нейроэндокринной и эндокринной системы (диэнцефальный синдром, несахарный диабет, транзиторный несахарный диабет, транзиторный гипопаратиреоз, инсулинозависимый сахарный диабет, поликистоз яичников, эутиреоидная струма, диффузный токсический зоб, субкомпенсированный гипокортицизм). У детей с синдромом Платтера, отмечается тенденция к высоким показателям роста и слабой пигментации кожи, что может также указывать на дисфункцию гипоталамо-гипофизарной системы. Кроме того, у этих детей существует ряд симптомов, которые мы назвали торакальными и к числу которых относится синдром сдавления средостения. Этот синдром мы наблюдали только у детей первых 2–2,5 мес жизни при значительном увеличении тимуса.
Все исследователи, занимавшиеся изучением особенностей лиц со стойкой тимомегалией (а основную группу в них, вне всякого сомнения, составляют дети с синдромом Платтера), обращали внимание на их пониженную физическую активность.
При специальном опросе 72 пациентов в возрасте 10–24 лет с синдромом Платтера у 46% из них было отмечено негативное отношение к физическим нагрузкам, особенно к бегу на длинные дистанции и езде на велосипеде. Подростки и лица молодого возраста, способные передать ощущения, появлявшиеся при физической нагрузке, отмечали на ее фоне чувство слабости, ощущение нехватки воздуха.
Когда у 30 подростков с синдромом Платтера, не имевших органических заболеваний сердца, было проведено эхокардиографическое исследование сердца на аппарате 128 х Р — ACUSON (США) с определением массы миокарда левого желудочка сердца, у 27 из них индексированная (относительно массы тела) масса миокарда левого желудочка сердца оказалась достоверно меньше (р
Л. Г. Кузьменко, доктор медицинских наук, профессор
РУДН, Москва



